
At her first birthday, Emma Larson was not walking or standing, but neither are plenty of other kids at that age. She loved the bouncer her parents set up in their Long Island, N.Y., home, and she crawled with gusto. Then, at 13 months, Emma’s legs stopped working. Her mother, Dianne Larson, snaps her fingers and says, “It was like that.” Emma stopped bouncing. Her legs buckled when she pulled herself up to stand. The change in her crawling was subtler, but when her parents looked at an old video, the difference was obvious—Emma now covered less ground and struggled to hold her head up.
After a barrage of testing, in July 2014 the Larsons learned that Emma had spinal muscular atrophy (SMA), a potentially deadly neurodegenerative disease that strikes mostly children, robbing them of the ability to walk, talk and, in the worst cases, breathe. Her motor neurons were dying because of a severe lack of a protein called SMN (survival motor neuron) in her body. “You go through the darkest of dark periods,” Dianne says. But the family was determined to “go down swinging,” says Matt Larson, Emma’s father. “We were willing to do pretty much anything to combat this terrible disease.”
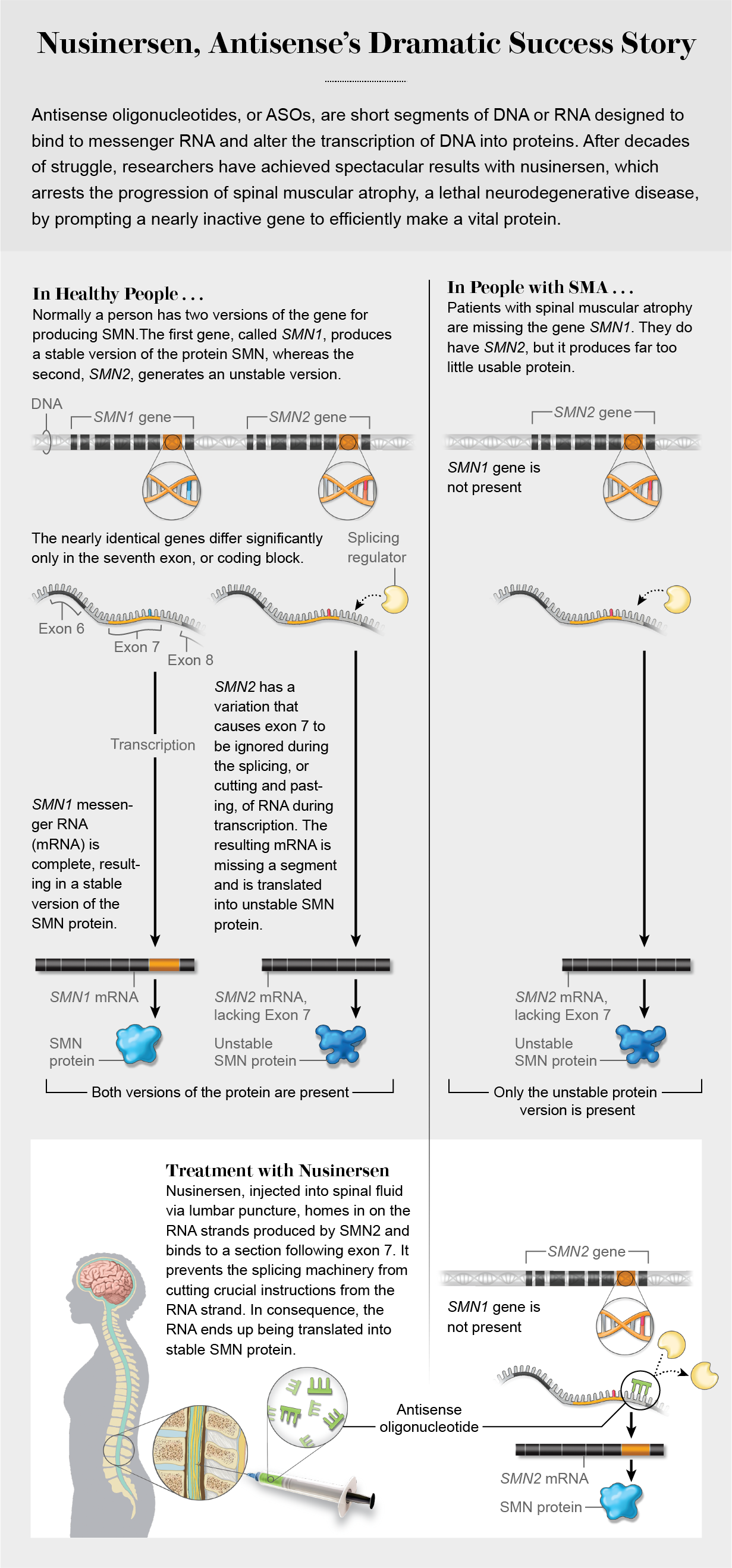
Not far from the Larsons’ home, at Cold Spring Harbor Laboratory, biochemist and molecular geneticist Adrian Krainer was engaged in the same fight. He had been investigating the genetic underpinnings of SMA since 2000 and knew the problem was a missing or mutated essential gene, SMN1. But he also understood that people carry an inactive and potentially salvageable analogue of that gene, SMN2. By 2004 he had joined forces with Frank Bennett of Ionis Pharmaceuticals to try to create a drug capable of altering SMN2 in SMA patients so that it could ultimately generate functional SMN protein, with the aim of ameliorating the progression of the disease. To that end, the researchers turned to something called antisense oligonucleotides.
First conceptualized more than 40 years ago, antisense oligonucleotides (ASOs) are short strings of chemically modified DNA or RNA (oligo in Greek means “few,” and nucleotides are the structural units that make up DNA and RNA). ASOs are designed to home in on the RNA strands produced by a problematic gene and alter the gene’s expression. That is, the ASOs bind to a section of the targeted RNA to produce (or, in some cases, stop the production of) proteins whose absence (or presence) causes an ailment. For decades scientists had labored to prove that this strategy could yield a drug capable of treating or preventing disease, but they found such serious problems with toxicity and delivery that many abandoned it. Yet the few researchers who pushed on overcame the obstacles just in time to benefit from the detailed information about genetic diseases revealed by the genomics revolution. “Antisense is tailor-made for diseases that have a genetic cause,” says Brett Monia, who took over as CEO of Ionis in January from founder Stanley Crooke. “It’s the epitome of precision medicine.”
Krainer, Bennett and their colleagues called their SMA drug nusinersen. When injected into cerebrospinal fluid, it coaxes the inactive motor neuron gene to make SMN. With Biogen, they began testing the drug in human clinical trials in 2011. The Larsons enrolled Emma the day she was eligible: her second birthday. By then she could no longer crawl at all. Her first dose, in March 2015, was followed by two more doses in quick succession.
In May 2015 Dianne was in her bedroom while Emma was in the den nearby. “I hear her calling me, and it’s getting closer and closer,” Dianne remembers. “Next thing you know she’d crawled from the den all the way to my bedroom.” Dianne asked herself, “Did I just see this?” She picked her daughter up and carried her back down the hall. Then she returned to the bedroom and called, “Emma, come here.” The little girl crawled into her mother’s arms. Weeping, Dianne thought, “We’re on to something here!”
Indeed, they were. The clinical trial for nusinersen proved so successful that it ended a year early. The U.S. Food and Drug Administration approved the drug, under the brand name Spinraza, in December 2016. More than 8,400 patients in 40 countries are now taking it. Twenty-five newborns with the most severe SMA mutation were given the drug at birth. They are four years old now—and developing normally. “If I had done nothing else but develop Spinraza, it would have been enough,” Crooke says.

But Spinraza is also exhibit A in support of the argument that ASOs are finally achieving their full potential. It is the first ASO to boast such dramatic results and commercial success. The drug earned Krainer and Bennett the multimillion-dollar 2019 Breakthrough Prize. It also put in reach a tantalizing set of neurological targets such as Huntington’s disease and amyotrophic lateral sclerosis (ALS). “We discovered the genetic basis for most of these diseases back in the 1990s,” Bennett says. “It’s taken us 25 years to translate these really important scientific discoveries into potential therapeutics. [With Spinraza,] it was breathtaking almost to realize that we had a technology that could have such a broad impact on patients who have no therapies available to them.”
Like long-distance runners who have been training at altitude, antisense scientists have put in hard miles to optimize the chemistry and delivery of oligonucleotides. Now they are at sea level and sprinting. More than 100 drugs are in the development pipeline for everything from Alzheimer’s disease to hypertension. Not all will reach the finish line, but, including Spinraza, eight have been approved so far in the U.S. and Europe, all for rare diseases. Drugs for Huntington’s and ALS are in the final stages of clinical trials. In a historic first, a doctor at Boston Children’s Hospital created a custom antisense drug for one little girl with an ultrarare disease in less than a year. “People have been talking about biologic therapies for 30 years, and what’s extraordinary is it’s starting to happen,” says neurologist Robert Brown of the University of Massachusetts Medical School, who is a leader in ALS research. (Biologic drugs are those made from living organisms.) “This is a true game changer.”
Sense and Antisense
DNA provides the basic blueprint for life, but it has to be read and translated into action through the production of proteins, which carry out most of the work in the body. Because the instructions encoded in DNA are so critical, the process of translation has protective mechanisms built into it. There is a lot of repetition, beginning with the two strands of nucleotides that zip together to form DNA’s double helix. One serves as a template, laying down sequences of the four bases that make up DNA molecules: adenine (A), thymine (T), guanine (G) and cytosine (C). The other strand reads that template and lays down a complementary set of bases. Each base on a strand is always positioned opposite its specific partner: A always pairs with T, and C with G. To ensure accuracy, the RNA only ever encodes the instructions in the nontemplate strand for the creation of proteins. Biologists call the two strands by a variety of names, including sense and antisense, which gives the ASO technology its name.
Occasionally the end results—the proteins—do not come out right. They can be overproduced or underproduced, resulting in disease. Small-molecule drugs, which make up the majority of pharmaceuticals on the market, target the proteins associated with diseases. Monoclonal antibodies, the other major class of drugs, generally bind to proteins and stimulate a patient’s immune system to attack them. In contrast, the aim with antisense drugs is to disrupt the process earlier. They are designed to replace faulty RNA during the transcription process by snapping into place according to the standard base-pairing rules and thereby tweaking protein production.
A parallel effort has focused on what is known as RNA interference, or RNAi. This technology was discovered just when antisense had been given up for dead, so its proponents avoided the term, but the drugs derived from the two approaches are related. “I think of antisense as the genus and RNAi as a species,” Bennett says. The difference is that RNAi drugs have two strands, whereas ASOs have only one. But any chain that is short—usually 15 to 20 nucleotides—is considered an oligonucleotide.
The versatility of oligonucleotide drug technology derives from the way it separates two critical elements: the platform or molecular properties that determine drug delivery and distribution into tissues, and the sequence of bases necessary to target a specific gene. Different sequences of bases make the information contained in the drugs distinct, but antisense drugs with the same chemical modifications tend to behave in similar ways in the body. “That’s what allows us to move quickly once a platform is established to deliver to a tissue of interest,” says Jonathan Watts, a nucleic acid chemist at the University of Massachusetts Medical School. “By shuffling the sequence of bases, we can dial in a totally different target using the information from a genome-sequencing experiment of a patient with a rare disease or from the genome databases. Being able to use that information intuitively and rationally is very powerful.”
A Long-Distance Run
The idea of using genetic information to make a drug that could bind to RNA has been around since 1978. But there were a host of unanswered questions: How do you make an oligonucleotide into a drug? Why would binding to RNA produce an effect? Nonetheless, the idea was intriguing enough to Crooke that in 1989 he left his position as head of research and development for SmithKline (now GlaxoSmithKline) to establish a company dedicated to the development of antisense technology. He was joined there by his wife, Rosanne, also a pharmacologist, and by colleagues, including Bennett and Monia. (Originally called Isis, the company eventually changed its name, for obvious reasons, to Ionis Pharmaceuticals.)
A handful of other companies started up to pursue antisense around the same time, but one by one most abandoned the hunt. For a time it did seem that the problems of toxicity, off-target effects and a lack of potency might not be overcome.*
But Crooke and his colleagues doggedly solved the scientific problems one at a time. A long, high wall of patents at Ionis’s headquarters near San Diego attests to their work. First they had to develop the necessary chemistry. For example, by modifying a key position (2′) in ribose sugar in the RNA and DNA of ASOs, they were able to enhance the affinity of the ASOs for RNA receptors, thereby dramatically reducing the necessary dose. Other chemical modifications improved safety and tolerability. They also found that the drugs were not taken up into tissue when delivered directly into cells in culture, but Ionis scientists made the leap to testing the drugs in animals anyway. Monia, who ran drug development for Ionis, vividly remembers the moment when he looked at a chemical test he was using to measure levels of a specific RNA and saw almost no trace of it—the drug had entered cells in most tissues, and they had successfully knocked down the RNA’s expression.
Time spent working on cancer did not prove all that fruitful, Bennett says. (Promising, more carefully designed experiments are in the pipeline, however.) What did work were drugs with specific targets, usually for rare diseases, for which proof of concept is easier to establish. The earliest ASOs were for diseases of the eye and, later, the liver, where uptake works particularly well. The drugs were effective, but they were ultimately not commercially viable, because better solutions came along.
The newest oligonucleotide drugs are designed to tackle rare diseases. One is Exondys 51, which targets Duchenne muscular dystrophy, a severe, progressive degenerative disease caused by mutations in the gene that produces the protein dystrophin. Annemieke Aartsma-Rus of Leiden University Medical Center in the Netherlands, who is president of the Oligonucleotide Therapeutics Society, is an expert in Duchenne and helped to develop the drug. It has been less spectacular than Spinraza, but on the strength of early results showing increased dystrophin levels, the drug received accelerated regulatory approval. The company marketing it (in which Aartsma-Rus has a stake) will need to show by 2021 that it makes a meaningful difference in how a patient functions.
The first RNAi drug, Onpattro, made by Boston-based biotech company Alnylam Pharmaceuticals, was approved in 2018 for treating a hereditary form of nerve damage. An approved Ionis ASO drug called Tegsedi treats the same thing. The focus now for all oligonucleotide therapies is delivering more drug more productively to more parts of the body. “A lot of people were in wait-and-see mode,” Aartsma-Rus says. “They now see that if they don’t start, they’ll have missed the boat.”

Hope for the Brain
For a long time antisense companies largely ignored neurological targets because oligonucleotides generally do not cross the blood-brain barrier. But Bennett thought that delivering them directly to the cerebrospinal fluid via lumbar puncture might work. He pushed a skeptical Crooke to let him try. “I had a lot of reservations, but the idea is to say yes,” Crooke says. “‘No’ never made a drug, and ‘no’ never made anybody better.” They started exploratory studies with a mouse model of Huntington’s, an obvious candidate for ASOs because it is directly linked to a specific mutation. People with Huntington’s carry a repeated sequence of a triplet of base pairs, CAG, that results in toxic levels of huntingtin protein and causes the progressive breakdown of brain cells. In mice, Bennett and his colleagues found that they could reduce levels of the mutant protein. “The mice actually improved,” Bennett says.
Meanwhile Krainer was investigating SMA. Others had discovered that healthy people have two versions of a critical motor neuron gene, SMN1 and SMN2, but the latter makes very little functional SMN protein. People with SMA do not have a functional SMN1 gene, and their broken copy of SMN2 cannot do the job itself. Stretches of DNA include both “exons,” the coding sequences that are expressed (hence the “ex” in their name), and “introns,” the noncoding stretches between exons. A process called RNA splicing joins the exons together and discards the introns. The SMN2 gene had a variation that rendered it inactive by causing a particular coding chunk, exon 7, to be ignored. Krainer and Bennett surmised that an ASO could force that instruction chunk to be included. By 2008 they had shown that the ASO they had created worked in mice by fixing the splicing defect. The clinical trials in humans followed.
“This is what’s called a disease-modifying therapy,” Krainer says of Spinraza. “It isn’t just dealing with some symptoms. It’s getting at the root cause of the disease and changing its course.” Early intervention is critical. A person with symptoms, such as Emma Larson, has already lost some motor neurons, which cannot be restored. But the treatment can prevent the remaining neurons from dying off and bring improvements in motor function. The success in treating infants has led to a push for newborn screening for SMA, which now occurs in 16 states. “The closer you start the treatment relative to birth or disease onset, the more you can achieve,” Krainer says.
Spinraza’s clinical success showed that, contrary to expectations, antisense therapy could be particularly effective against brain diseases. Neurological targets have “become the low-hanging fruit,” Aartsma-Rus says. Several ASO-based therapeutics are in development for Huntington’s, for example. One, known as RG6042, developed by Ionis and Roche, is in a phase 3 clinical trial. Earlier safety and tolerability studies showed that it is possible to lower levels of mutant proteins, says Scott Schobel, clinical science leader of the global Huntington’s ASO program for Roche, but “what now is the clinical import of that?” The current trial should answer that question. “We would consider even a 30 percent slowing of decline a victory,” Schobel says. That would amount to giving patients three to four months back out of a year while they are still functional.
Also known as Lou Gehrig’s disease, ALS is more complicated because at most 10 percent of cases have a clear genetic cause that runs through families. The most common inherited form is caused by a mutation in a gene called C9orf72; another gene, SOD1, causes about 20 percent of familial cases. Those make up a fraction of all cases, but the promise of antisense has injected new hope where there was previously little. “My mood is sky-high,” says ALS researcher Brown, who led the team that identified SOD1 in 1993. Clinical trials for antisense drugs to treat both the C9orf72 and the SOD1 forms of the disease are underway. The drugs have proved safe and tolerable and suppress the activity of mutant proteins.
Part of what has clinicians such as Brown so excited is that antisense has also made it possible to develop drugs for individual patients. A young Iowa woman named Jaci Hermstad, who has a very rare form of ALS caused by a mutation in a gene called FUS, began taking a drug tailor-made for her in the summer of 2019. So far she is tolerating it well, and there have been small improvements, such as her regained ability to move her arm.
A Drug for Mila
A drug for just one person was science fiction until neurologist Timothy Yu of Boston Children’s Hospital created a drug in less than a year (record time) for Mila Makovec, now nine years old. Mila has an ultrarare condition called Batten disease, which is really a family of disorders in which mutations cause buildups of proteins and lipids in cells. Children with Batten rarely survive into adolescence.
Like many people with Batten, Mila was unusually well coordinated and verbal early on. But at three, her toes started turning inward. Between four and five, she got clumsier and started losing her vision. Doctors at Children’s Hospital Colorado eventually connected Mila’s symptoms with one gene mutation for Batten that she carried.
But Batten requires two gene mutations. Mila’s mother, Julia Vitarello, went looking for someone who could fully sequence Mila’s genome to confirm the diagnosis. She and Mila’s father also wanted to know whether their younger child, Azlan, was at risk. In January 2017 her plea reached Yu’s wife via social media.
Yu’s team did the sequencing and found the missing second mutation. It was caused by a jumping gene, or transposon, a sequence of nucleotides that replicates and moves to a spot in the genome where it does not belong. The discovery meant Azlan was safe. It also gave Yu an idea: it might be possible to create a drug for Mila. “We realized we could pull the Spinraza trick,” Yu says. “But instead of using an antisense oligo to force an exon that was being ignored to be included, we were using an antisense oligo to shut down an exon that was getting in the way.”
After several pharmaceutical companies demurred, Yu oversaw manufacture of the drug himself. Some of the $3 million Vitarello had raised in search of a cure went to the project (she prefers not to specify how much). Yu called the drug milasen, for the only patient who would receive it, and Mila got her first dose in January 2018. By then she was blind and having seizures 20 to 30 times a day, some lasting for several minutes. The damage already done to Mila’s body cannot be repaired, but with treatment her seizures soon eased. After four or five months, they were lasting only a few seconds rather than minutes. Vitarello says that recently, with her help, Mila even walked up stairs with alternating feet.
When Yu reported Mila’s story in the New England Journal of Medicine late in 2019, it made headlines. It also raised concerns about the cost and the ethics of developing a drug for one person. (Both Yu’s institutional review board and the FDA approved milasen.) Bioethicist Sara Goldkind, a former FDA staffer, as well as a consultant on rare disease programs and an adviser on milasen, says that process is critical in such an unusual situation. Tests for safety and effectiveness must still be done, but there are also many mitigating circumstances—these are rare, deadly and rapidly progressing diseases with no treatments—that might allow the FDA to rely on a single adequate and well-controlled study instead of the two usually required. “There needs to be some flexible thinking in terms of how the regulations are applied,” Goldkind says.
Crooke, who has stepped back from running Ionis day-to-day, set up a foundation to support the development of customized antisense drugs for ultrarare diseases affecting too few people to be viable commercially. Vitarello and Yu, too, hope to make personal treatments available to all children like Mila. One of the great advantages of antisense is that such individualized drugs can be created not just quickly but also relatively inexpensively, despite the considerable sums spent on Spinraza and milasen.
Emma the Flamingo
Like Mila, Emma Larson was not cured. The neurons she lost have not been replaced, and she has skeletal changes that are likely to be permanent. At the Larsons’ home, there are wide expanses of uncarpeted wood floor, the better for Emma, who just turned seven, to zoom around in the wheelchair she calls her race car. She is in first grade, and her favorite part of the day is recess, when she likes to play on the slide and the seesaw.
When the wheelchair is parked, her parents carry her from room to room. She crawls around her playroom to show off her favorite toy, a Polly Pocket Mall. But with her walker and braces attached to a pair of sparkly pink sneakers, Emma can take a few steps under her own power. And in the dining room, with one hand on the table, she stands on the bench along the wall like a flamingo and cries, “Hey, look, standing with one leg!”
Life is still hard, the Larsons admit, but they no longer despair. They hope Emma can live independently. And they are thrilled that the newborns on Spinraza are doing so well. “That made my heart full,” Dianne says as her eyes well up. “In some regards, it’s a little late for Emma, but she helped pave the way for those little babies.”
Credit: Mesa Schumacher
*Editor’s Note (4/17/20): This paragraph has been edited after posting to remove an erroneous reference to one of the companies pursuing antisense therapy.

