
It was the middle of the night in Jerusalem, and we were watching mice swim. The year was 1994, and the two of us were crouching over a pool of cold water in a laboratory at the Hebrew University. The room was chilly, our hunched backs ached, and we had been repeating this routine over many nights, so we were tired and uncomfortable. So were the mice. Mice really dislike swimming, especially in cold water—but we wanted to stress them out.
We humans were on the night shift because both of us had other things to do during the day. Kaufer was working on a doctorate in molecular neurobiology, and Friedman was an Israel Defense Forces physician and was often on call. What brought us together with the mice every evening was an attempt to understand a medical mystery: Gulf War syndrome. After the conflict ended in 1991, there were an increasing number of reports of soldiers from the U.S.-led coalition who were afflicted with chronic fatigue, muscle pain, sleep problems and cognitive deterioration, and those soldiers were hospitalized at higher rates than nondeployed veterans. Some doctors suspected that pyridostigmine, a drug that had been given to soldiers to protect them from chemical weapons, could cause these ailments if it made it into their brains.
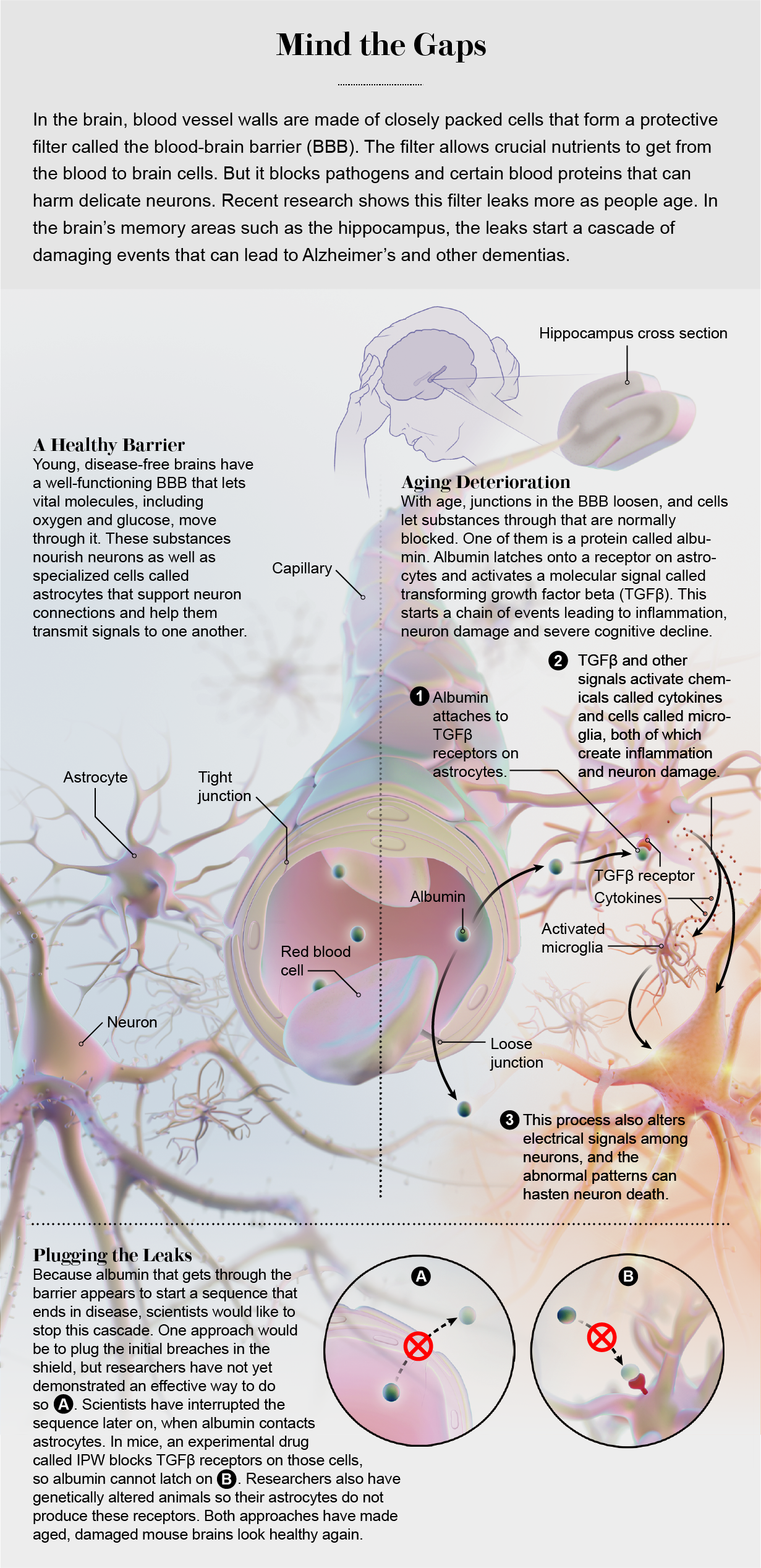
There was a big problem with this theory, however: pyridostigmine in the bloodstream was not supposed to reach the brain. Blood vessels that course through this vital organ have walls made of specialized cells, packed very closely and with abilities to control what can get in and out. They form a shield that keeps toxins, pathogens such as bacteria, and most drugs safely within the vessels. This structure is called the blood-brain barrier, or BBB for short, and the drug should not have been able to pass through it.
Unless, however, the barrier was not intact. We wondered whether the physical and mental stress of combat might somehow trigger leaks in the shield. The swimming mice were our way of testing whether stress led to damage. When the swim session was done, we pulled each mouse from the pool and injected a drop of blue dye into one of its veins. Then we waited as the dye passed through its body, gradually turning the mouse blue. If the BBB was intact, the brain should have remained its normal pinkish-white color. We euthanized the mice so we could take a look at their brains under a dissecting microscope. Over several nights we had tried various lengths of swim time, but we had not seen any changes.
But on this night, after two dips in slightly colder water, things looked different: the brains had a strong blue tint! Lab work is usually tedious, and success is often subtle, but this time we were jumping up and down and hugging each other, giddy with excitement. Our weird experiment had worked. Stressful situations could make the BBB spring leaks. With our mentor, neuroscientist Hermona Soreq, we went on to show that such changes let in pyridostigmine and altered brain cell activity. We published these results in 1996 in Nature Medicine and in 1998 in Nature.
A quarter of a century later we can say that looking at these blue brains turned out to be a defining moment for both our careers, as well as the beginning of a lifelong friendship and scientific collaboration. Discovering the telltale blue tinge was the first step on a path that, over many years, led us to probe more and more deeply into the connection between other brain diseases and flaws in the organ’s protective shell. Today pyridostigmine penetration is an important hypothesis for the cause of Gulf War syndrome, although there are other candidates. And our investigations have linked BBB damage—caused by aging or injury in addition to acute stress—to several more familiar illnesses: Alzheimer’s and related dementias, epilepsy and traumatic brain injury. In two papers published in 2019 in Science Translational Medicine, we demonstrated that as people get older, this shield loses integrity and starts leaking, allowing blood proteins into the brain that normally do not get there. These proteins in turn activate a cascade of events among brain cells that can produce some of the most notable and widely seen changes associated with aging and illness: inflammation, abnormal neuron activity and cognitive impairment.
The cause-and-effect connection looks especially strong because stopping the reactions set off by these leaks actually reverses signs of disease, at least in rodents. In older mice, we can abolish the inflammatory fog with a targeted drug that protects brain cells from being irritated by blood proteins or by making genetic modifications that prevent those cells from releasing inflammatory molecules. Within days of the treatment the aged brains of these mice started to function more like young brains. Abnormal electrical activity subsided. Markers of inflammation dropped to low levels. When placed in mazes, the animals made their way through as quickly and accurately as young mice did. We cannot try the same experimental brain modifications in humans; it is not ethical. But we have been able to use imaging techniques to compare the brains of people with Alzheimer’s with those of healthy people. The images show excessive and progressive BBB leaking in those with the disease, as well as other features of the illness-related cascade.
We do not yet know whether a damaged barrier is truly responsible for Alzheimer’s or other brain illnesses. It could play a contributing role along with other causes, including genetics and a variety of cellular problems that have been observed in aging brains. Or it could be collateral damage. And experiments in mice often do not pan out in people. But right now the long-standing dominant theory for Alzheimer’s—that it is triggered by a buildup of a protein called beta-amyloid in the brain—is looking less convincing than ever. Numerous experiments have reduced levels of this protein in the brain, yet the disease and associated mental decline in people remained unaffected. Drugs that target beta-amyloid have failed to help. Given that there are now 50 million people worldwide with dementia and another 10 million diagnosed every year, according to the World Health Organization, many scientists say it is high time to consider alternative explanations. If flaws in the brain’s protective shield start a chain of events that leads to disease—a chain that experiments suggest can be blocked to restore brain health—it is a path of investigation worth pursuing.
Flaws in The Wall
With “barrier” in its name, the BBB sounds like a wall around the brain, but it is really more like a distributed filter. Our body’s control center gets 15 to 20 percent of the oxygen-rich blood pumped out by the heart, delivered by an intricate mesh of blood vessels. They look different than vessels in the rest of the body, with walls made of tightly packed cells with specific molecular transport systems that form a semipermeable filter. Networks of brain cells need a carefully controlled environment to function, so this filter lets molecules such as oxygen and glucose get through but blocks blood proteins, certain ions, immune system cells and pathogens. This protective mesh extends throughout most areas of the brain, from the outer layers of the cortex, where higher-order cognition occurs, to deep places such as the hippocampus, which regulates memory storage. Problems with the filter can therefore lead to all kinds of neurological difficulties.
Back in the 1990s, as we were completing our initial work on Gulf War syndrome, we knew that other researchers had noted BBB damage in some brain disorders, including Alzheimer’s. But we did not know whether this problem was a cause or an effect or how leaks in the shield get started and what they might do to alter brain function. We did, however, want to find out.
After our time working in Jerusalem, Kaufer went to Stanford University for her postdoctoral fellowship, and Friedman continued his medical training in Israel, specializing in neurosurgery. But time and distance did not let us forget. On a vacation together with our families, sailing between Greek islands, we caught up. Kaufer was learning more about how stress affects the brains of mice in work at Stanford. Friedman, in his own practice, was reaffirming the early observations from other researchers who saw flawed BBBs in many patients suffering from very different neurological conditions. Just what was the damaged barrier doing?
We began to figure out the answer to this question in the mid-2000s, when we got the chance to work in Berlin with the late neuroscientist Uwe Heinemann of the Institute for Neurophysiology, part of the Charité Center for Basic Medicine. Heinemann opened his lab to our next key experiment. We wanted to observe brain function directly after the BBB started to malfunction, so we gave rats a chemical that essentially poked holes in the barrier and then dissected their brains. We kept the brain slices alive in nourishing fluid and used an electrode to record the electrical signals that the cells used to communicate with one another.
The first few days were boring. The neurons were giving off signals one after another in staccato, irregular patterns, “talking” as if nothing unusual had happened. We almost decided to give up. Then, on the fifth day, the cells’ chatter patterns changed. More and more neurons started to pulse together in synchrony. After a full week we nudged them with a small signal from an electrode, mimicking a brief electrical message within the cerebral cortex. This nudge produced a storm of cells firing together, similar to what is observed in people and animals with epilepsy.
We think what happened with these cells is analogous to generating a Twitter storm. Imagine that you created a Twitter account today and tweeted some sensational statement. You would probably get a very small response because you would not have many followers. If in the next few days you built a bigger network of followers and tweeted again, however, the same statement would be likely to be retweeted, recruiting more followers who would also retweet it, eventually leading to a storm of tweets on the social media platform. Similarly, when we disrupted the BBB, neurons in the brain were not discombobulated right away, but after they had spent a week building a new network of connections, a small jolt prompted a big electrical storm. These patterns, which we call paroxysmal slow-wave events, are similar to activity seen by other scientists in the brains of people diagnosed with Alzheimer’s and with epilepsy.
This storm happened only after we mimicked a BBB leak. Without one, our brain slices were untroubled by any electrical tempests. So we hypothesized that there was some element from the blood that was reaching these neurons to trigger the brain reaction. We tested this theory in a young, healthy rat with a normal BBB by injecting blood directly into its brain—bypassing the barrier—and monitoring electrical activity. It took several days, but again the storm built and exploded. Clearly, it had something to do with the blood. But blood is a complex fluid containing many different kinds of cells and proteins, so we set off on a painstaking filter-and-trap expedition to isolate the culprit. Eventually we found one blood protein that created the disturbances: albumin.
The Start of Trouble
We were not thrilled with our catch. Albumin is very common and is involved in many bodily functions, so it was hard to isolate what it was doing in this situation. We would have preferred a rarer component. But albumin was what we got, so we dug in. Kaufer moved to the University of California, Berkeley, to run her own lab, and Friedman started his, first at Ben-Gurion University of the Negev in Israel and later at Dalhousie University in Nova Scotia. We planned a joint, long-distance series of experiments over several years to delineate the steps from BBB disruption and albumin leakage to the appearance of neurological disorders.
The first thing we learned was that when albumin gets into the brain, it appears to stimulate astrocytes, key brain cells that provide structural and chemical support for neurons and their connections. When albumin contacts an astrocyte, it binds to receptors that usually serve as docking stations for a molecule called transforming growth factor beta (TGFβ). Among other things, TGFβ activates astrocytes and sentinel cells called microglia to start inflammation. Normally, localized inflammation is the brain’s way of limiting damage by destroying malfunctioning cells in a targeted assault. But if albumin continues to seep in, the astrocytes and microglia get hyperstimulated, and too many damaging chemicals get released, including an overabundance of TGFβ. Lots of brain cells get hurt, and key neural circuits are modified or weakened, so their functions deteriorate. Often doctors have observed this same destructive cascade in patients after traumatic brain injury; sometimes it leads to epileptic seizures.
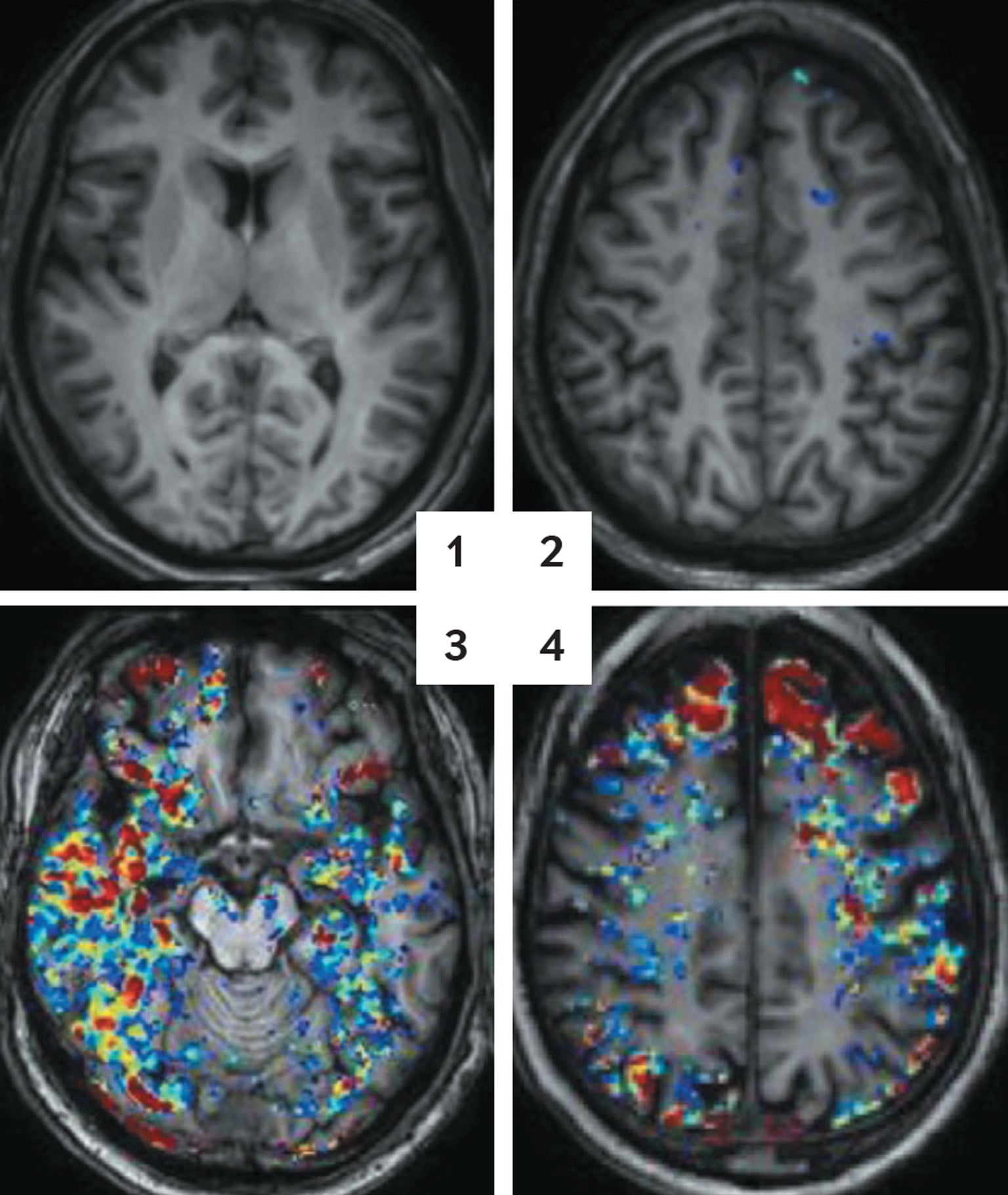
The sequence shows up in the aging brain as well, as we learned by looking for it in mice. The animals typically live a bit more than two years on average. We allowed a colony of mice to age peacefully and looked inside their brains at various points. Albumin, we saw, was not in the brain at all in younger mice, but it began to show up in middle age. The effect was modest at first, but there was a clear decline in the integrity of the barrier, and it got worse as the mice got older. The affected mice also had more trouble remembering their way through mazes than did their younger and relatively albumin-free counterparts.
When the albumin showed up, other experiments showed us, TGFβ started to get active. We stained the brains in ways that highlighted the activated form of the growth factor as well as the astrocytes that produced it. The inflammation related to TGFβ always started after albumin appeared, and it got worse as more of the protein leaked in. This pairing was especially abundant in the hippocampus, a brain area that is a key component in memory regulation.
Within the past five years or so we have been able to provide good evidence that this same process happens in people. We used tracer molecules to tag signals of barrier leakage in people in their mid-20s to mid-70s. With magnetic resonance imaging, we could see that the degree of these drips increased as people got older. Other researchers, such as Berislav V. Zlokovic of the University of Southern California Keck School of Medicine and his colleagues, used slightly different imaging methods to show barrier deterioration in aging people with cognitive impairment. In our work, we added autopsies of a separate group of people and showed that heightened albumin levels accompanied greater amounts of TGFβ, always in astrocytes. These concentrations were higher in older people and heightened as well in people who had died of Alzheimer’s when compared with those without the disease.
Brain Rejuvenation
Then we reversed the deterioration in mice. We could not stop albumin from starting to seep through the BBB, but we could block the TGFβ cascade that came after the leaks. We developed a group of mice in which we genetically cut out the portion of DNA that tells astrocytes to produce TGFβ receptors, eliminating that feature from the cells. When the mice were still relatively young, we implanted a tiny pump in their brains that injected albumin. We did the same thing to a group of young, normal mice. Then we put both groups into a tricky water maze. (Watching mice swim seems to be a recurring theme with us.) The mice with receptors had a lot of trouble. But the animals without receptors swam the maze like young, healthy mice—speedily and accurately—and when we changed the maze configuration, they learned the new route, too. When we looked at their brains, we saw low levels of both inflammation and abnormal electrical activity.
This was really very encouraging. But for people, the option of knocking out a gene for a brain feature will not be available therapy any day soon. There is, however, another form of medicine. Barry Hart, a medicinal chemist at Innovation Pathways, a start-up drug company in Palo Alto, Calif., had designed an anticancer drug that specifically blocked the activity of the TGFβ receptor. Hart contacted us and suggested that we try the drug, called IPW, on our mice. (The three of us have since formed a company to further develop the medication.)
When we gave the drug to middle-aged mice—the ones that were starting to show albumin leakage—we learned that it made their brains look young again. TGFβ activity dropped to levels seen in youthful mice, markers of inflammation went way down, and abnormal electrical activity and seizure susceptibility diminished.
But the big surprise came when we tested actual behavior and cognition. We set up another maze, and this time we ran older mice through it. Some of the aged animals were treated with IPW, and some were not. We did not predict a lot of improvement, because we thought irreversible damage had already been done. (Our mice without the TGFβ gene had been spared the long months of deterioration inflicted by the inflammatory cascade, but these animals had not.) Within days, however, the treated mice were almost as good at learning the maze as rodents half their age. The untreated mice just shambled along as usual. Moreover, the mice that got IPW showed no sign of the “Twitter storm” effect that we typically see in humans with Alzheimer’s or epilepsy and not much evidence of inflammation. It was as if an inflammatory fog had lifted, allowing the brain to regain its youthful abilities. These, along with the studies of human brains, are the results we published in 2019 in Science Translational Medicine.
The maze outcome was so unexpected, even to us, because, like most people, we had considered aging damage as a one-way trip—deterioration that cannot be undone. That is probably the case for major brain trouble, such as the havoc that occurs in Parkinson’s disease or in advanced Alzheimer’s after so much beta-amyloid has accumulated that it kills off swaths of neurons and other cells. But our intervention research may indicate that in the absence of a lot of cell death, the aging brain has a hidden capacity to rebound from some types of insults.
And our findings have implications for acute injuries as well, not just gradual deterioration. Treating rodents with IPW after concussions or traumatic brain injury alleviated the inflammation, seizures and cognitive decline that they developed. Animals that got a neutral placebo drug were not helped.
Fixing the Damage
The world population is aging, and the number of people with dementias and Alzheimer’s is on the rise. We are aging, too, so this is personal. Both of us are about 50 years old, and our dinner conversations with friends often revolve around our concerns with our aging bodies (some of us used to run marathons and now cannot even finish a Zumba class) and brains (Kaufer cannot remember the names of the parents in her daughter’s class at school). Neuroscientists have a poor understanding of the early triggers of this transition from a young, healthy brain to an old, dysfunctional one. Alzheimer’s and other neurological diseases of aging are complex and can have many causes.
Now a leaky BBB has to be considered as one of them. This barrier-breach theory provides a remarkably intuitive and straightforward new model to understand why the brain declines with age. And it is a model that gives us optimism: the results of our work strongly hint that the aging brain retains a capacity for reshaping and restoring itself, an ability that may be suppressed, but not irreversibly lost, by persistent leakiness and the ensuing chain of events.
The next step for us and for other scientists is to look for strategies and therapies to reduce barrier leakage. In the past, pharmaceutical research into the barrier focused on ways to increase permeability, not limit it, to get more drugs across it to treat brain tumors or infections. Our results show that it is time to flip the question: Can we come up with ways to stop the shield from degrading, stop harmful substances from getting across, or at least interrupt the fall of molecular dominoes if they do? There is a chance to do a lot of good for a lot of people if we can figure these things out.
 Rights & Permissions
Rights & Permissions


